第71回 6種の類人猿の完全なゲノム解読がなされる
1.はじめに
米国ペンシルベニア州立大学等の研究者らは、6種の類人猿のほぼ完全なゲノム解読を行い、本年5月、ネイチャー誌に掲載された(オンラインでの発表は4月)。これにより、各類人猿の特殊な染色体構造や遺伝子が明らかになったほか、いくつか意義深い発見があった。今回はこれについて、背景状況も含め、分析・考察を行う。

2.類人猿とは
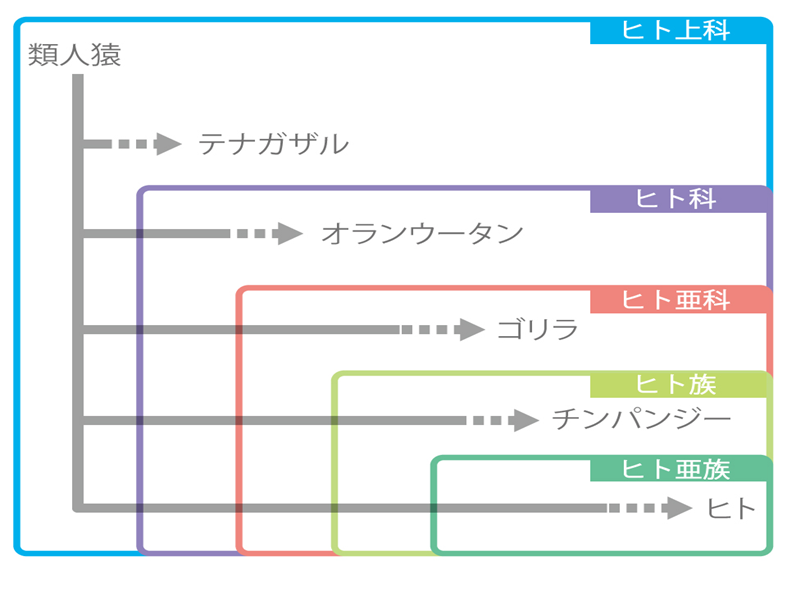
生物は学問上、ドメイン・界・門・鋼(類)・目・科・属(族)・種として分類される。種は一番下の単位であり、種が違うと普通は交接により子供を作ることはできない。我々ヒトは、真核生物ドメイン・動物界・脊椎動物門・哺乳鋼(類)・霊長目・ヒト科・ヒト属・ヒト(種)として位置づけられる。
このうち、ヒト上科に属するものは「類人猿」(ape)と呼ばれ、ヒト以外の大型類人猿(great ape)としてオランウータン、ゴリラ、チンパンジー、ボノボ、小型類人猿(lesser ape)としてテナガザルとフクロテナガザルがある。
なおヒトや類人猿を含む「霊長目」(primate:慣用名で「霊長類」と呼ばれる)のうち、ヒト以外を「サル」としていることが多い。
3.類人猿のゲノム解読の進展
ヒトゲノムは2003年に当初の解読が完了したが、それ以来、他の類人猿のゲノム解読によるヒトゲノムとの比較が次の目標の一つになっていた。なぜなら、そうした比較により、ヒトの脳の発達の解明やヒトに特有な疾患等の解明等が期待されたからである。
その後、チンパンジーは2005年、スマトラオランウータンは2011年、ゴリラは2012年、ボノボは2021年にそれぞれゲノム解読がなされてきた。
また、類人猿も含めた、広く霊長類全体について、中国科学院(CAS)傘下の昆明動物研究所(別サイトのこちらを参照)が主導する「霊長類ゲノムプロジェクト」と、イルミナ社が中心となって進めるプロジェクトにより、ゲノム解読が行われてきた。その状況についてはかつて本ニューズレターでも紹介しており(第27回 霊長類のゲノム解読)、2023年7月の段階で、前者は霊長類の14科38属をカバーする50種のゲノムが、後者は霊長類の16科全てと、属のうち86%をカバーするゲノムが解読されている。
しかし、これらの解読結果には問題があった。つまり、それらは完全なゲノム解読ではなかったのである。
高等動物のゲノムには、高度の反復性をもつ繰り返し配列が存在する。それは特に、染色体の中心部にあるセントロメアや、染色体の端にあるテロメアの部分に多く存在する。これらはそれぞれ独自の働きを担っている重要な部分であり、その反復配列のためか、これらの部分ではしばしば通常の二重らせんとは異なる異常な構造が形成される。そのような構造は「非B-DNA」と呼ばれる。
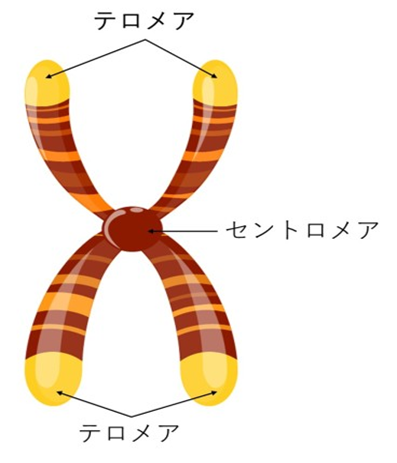
しかし、従来のゲノム解読では、この繰り返し配列の並びを正確に決定することはできなかった。
ゲノム解読を行うためには通常、染色体DNAを短い断片にばらばらに切断し、それぞれの断片毎に塩基配列を決定する。そして各断片の重複部分(のりしろ)を使って各断片の並びを決定する。
ところが繰り返し配列の一単位がこれら断片より長い場合、断片どうしの位置関係が分からなくなる。特に従来型の次世代シーケンサーは一般に、読み取れる断片の長さが数十~数百塩基対と短いため、繰り返し配列部分の解読には向かなかった。
このため、これら繰り返し配列部分が未解読であることで、それらが造る大きな構造の比較ができなかった。
4.ロングリードシーケンシング技術の発達
このような閉塞感を打ち破ったのが、数十万塩基対という長いDNA断片を読み取る、ロングリードシーケンシング技術の発達だった。特に、ナノメートル単位の穴(ボア)にDNA断片を通すことで、その微細な電流変化を読み取っていく技術が大いに進展し、読み取る長さや正確性を向上させてきた。
米国ヒトゲノム研究所(NHGRI)を中心とする研究グループ「テロメアtoテロメア コンソーシアム」(T2TC)は、この技術を用いて、2022年にヒトのゲノムを完全に解読した(第2回 ヒトゲノムの「完全な解読」)。
そして、この方法を利用することで、類人猿をはじめとする他の生物のゲノムも完全解読が行われることが期待された。
5.今回の研究の手法と成果
今回、T2TCの一環として米国ペンシルベニア州立大学の研究者らが解読したのは、チンパンジー(Pan troglodytes)、ボノボ(Pan paniscus)、ゴリラ(Gorilla gorilla)、ボルネオオランウータン(Pongo pygmaeus)、スマトラオランウータン(Pongo abelii)、フクロテナガザル(Symphalangus syndactylus)の6種のゲノムである。
彼らは、先述のロングリードシーケンシング技術と計算アルゴリズムを用いることにより、これまでシーケンシングが困難だったテロメアやセントロメア部分を含め、各類人猿について99%を超えるゲノムの塩基配列を決定した。これにより、種間のより正確な比較と、遺伝的変異のより深い理解が可能となった。
今回新たに解読された領域には、先述の非B-DNA構造を形成する配列が豊富に含まれ、これが遺伝子転写やDNA複製等の重要な細胞プロセスを制御する役割を果たす可能性が示された。
また、これらの領域から、特定の種や種群に特異的な新たな遺伝子や、多くのコピーが存在する遺伝子ファミリーが発見された。それが知能等人間特有の特徴を含む、種間で見られる違いに部分的に関係している可能性があることが示された。
繰り返し配列部分の比較によって、ヒトと他の類人猿は、ゲノムの他のどの領域よりも、最近になって生じた繰り返し配列において、その配列と構造の違いが大きいことも明らかになった。
さらに、本研究により、霊長類の種間の分岐時期の正確な推定値が示された。たとえば、ヒトとチンパンジーは約550万年前から630年前に分岐したと推定され、オランウータンは約1,820年前から1,960年前にアフリカの大型類人猿から分岐したと推定された。
6.おわりに
このように、類人猿の完全な形でのゲノム解読がなされることで、少なくともゲノムの世界では、必要となる情報が全て入手されることになる。そうすると、構造の違いも含め解析の精度はぐっと上がることになる。これにより、ヒトの脳等の発達・進化の理解、ヒト特有の疾患等のメカニズムや治療法の解明、さらに絶滅危惧種の類人猿を保護する取組み等にも役立つことが期待される。
今後の研究の進展は、比較対象となる生物を類人猿から霊長類、哺乳類等、各種生物に遡っていくことや、AI等も用いた解析技術の進展、さらに、RNA、タンパク質、エピゲノム等、他のオミックス技術の発展に依拠するところも大きく、引き続きフォローしていきたい。
参考文献
・D. Yoo et.al. (2025) “Complete sequencing of ape genomes”, Nature; Vol.641, 401-418
・L. Kuderna (2025) “Evolutionary insights from complete ape genomes”, Nature; Vol.641, 313-314
・“Complete genome sequences of six ape species unveiled” (2025/4/9) Penn State Univ. HP
https://www.psu.edu/news/research/story/complete-genome-sequences-six-ape-species-unveiled
・「これは「最古の人類」なのか…? 「チンパンジー亜属との分岐点」に生きた霊長類が、頭骨に残した「衝撃の特徴」と、謎」msn 現代ビジネスHP(図として引用)https://www.msn.com/ja-jp/news/opinion/%E3%81%93%E3%82%8C%E3%81%AF-%E6%9C%80%E5%8F%A4%E3%81%AE%E4%BA%BA%E9%A1%9E-%E3%81%AA%E3%81%AE%E3%81%8B-%E3%83%81%E3%83%B3%E3%83%91%E3%83%B3%E3%82%B8%E3%83%BC%E4%BA%9C%E5%B1%9E%E3%81%A8%E3%81%AE%E5%88%86%E5%B2%90%E7%82%B9-%E3%81%AB%E7%94%9F%E3%81%8D%E3%81%9F%E9%9C%8A%E9%95%B7%E9%A1%9E%E3%81%8C-%E9%A0%AD%E9%AA%A8%E3%81%AB%E6%AE%8B%E3%81%97%E3%81%9F-%E8%A1%9D%E6%92%83%E3%81%AE%E7%89%B9%E5%BE%B4-%E3%81%A8-%E8%AC%8E/ar-AA1otURE
・「染色体の図」沖縄大学院大学HP(図として引用)
https://www.oist.jp/ja/image/diagram-chromosome
ライフサイエンス振興財団嘱託研究員 佐藤真輔
