第90回 ゲノムの変異による影響を解明するAIモデルが登場
1.はじめに
グーグル・ディープマインド(Google DeepMind)社は、ゲノムの変異に応じてDNAがどのような機能を持ちうるかを予測できるような新たなAIモデルを開発し、本年1月、ネイチャー誌に掲載された。アルファゲノム(AlphaGenome)と名付けられたこのモデルは、遺伝性疾患のさらなる理解、より高度な遺伝子検査、新たな治療法の開発の指針等としての活用が期待されるが、今回はこのモデルの背景や特徴、意義等について分析・考察したい。

2.本モデルを巡る背景
(1)ヒトゲノムについて
基礎知識として、ヒトゲノムの構造や働きについての研究や開発の状況を、歴史とともに簡単に述べる。なお以下は著者の知見の範囲にとどまるため網羅的・体系的ではない。
ヒトゲノムについては、1991年から2003年にかけて世界各国の協力により行われたヒトゲノム計画により、初めて全ゲノムの解読がなされた。その後は次世代シーケンサーの急速な発達にも伴い、数多くのヒトゲノムが解析された。
こうした努力等により、ヒトゲノムを構成する約30億対のDNA塩基配列の中に、タンパク質になる遺伝子は約2万個~2万2千個存在することが分かってきた。さらに、疾病患者と健常者のゲノムの比較研究等により、各種遺伝子変異によるタンパク質の働きの変化についてもある程度の情報が得られてきた。
しかし、遺伝子はゲノム全体から見ると非常にわずかな割合である。
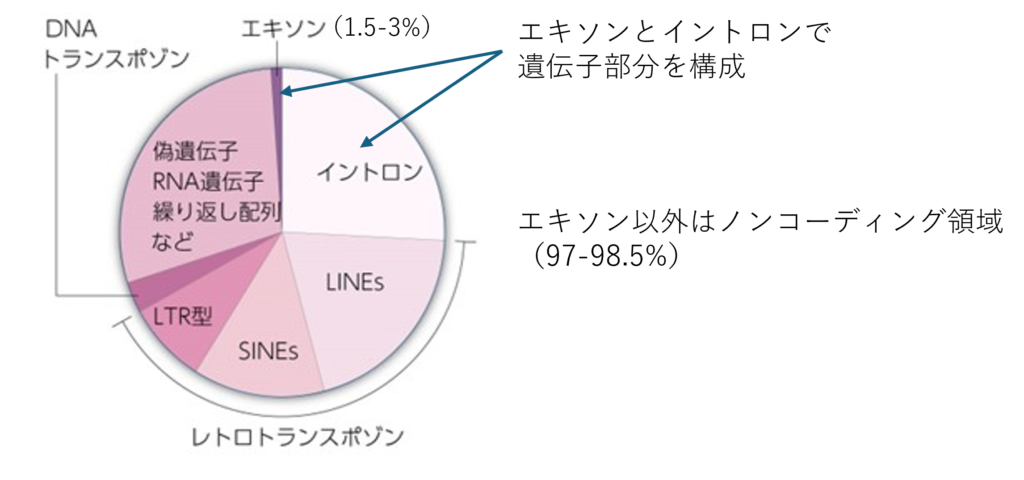
まず、遺伝子のある部分は、mRNAへの転写され、さらにタンパク質へと翻訳される「エクソン」領域と、mRNAに転写された後に切り出されて(スプライシングという)捨てられる部分である「イントロン」領域からなっている。このように、エクソン部分は実際にタンパク質をコードすなわちアミノ酸配列を指定する部分なのだが、これはゲノム全体の約1.5~3%程度にすぎないことが分かっている。
ではイントロンを含めた残りのゲノム領域、つまりゲノム全体の約97%〜98.5%を占める、いわゆる非遺伝子領域(ノンコーディング領域)はどんな働きをしているか。
このうち最も多く、全体の40パーセント以上を占めるのは「 レトロトランスポゾン 」 という、ゲノム上を動き回るトランスポゾンの一種である。レトロトランスポゾンは、かつて人類の祖先に感染したウイルスの遺伝子が、そのまま我々のゲノムに残ってしまったものと思われるが、現在のヒトではどんな働きをしているかよく分かっていない。また、既存の遺伝子によく似てはいるが、翻訳はされない偽遺伝子、転写されてRNAにはなるが、タンパク質に翻訳はされないRNA遺伝子、また、テロメアやセントロメア部分に多く存在する繰り返し配列等がある。
これら、ノンコーディング領域は、かつては全く機能を持たない、ジャンク(がらくた)だと考えられていた。しかし、研究が進むにつれ、これらの領域の多くの割合は、プロモータやエンハンサー等、遺伝子発現の調節に働く部分であったり、またRNAにはなるもののタンパク質に翻訳されない部分は、RNAそのものとして各種の働きを有すること等が分かってきた。つまり、ノンコーディング領域も、相当部分はジャンクではなく、それなりの役割を持つということなのである。
(2)各遺伝子の役割の解明
上述のように、これらヒトゲノムの配列が解読され、また、それぞれの遺伝子の働きも次第に分かってきた。ただしそれは、世界の研究者が、もともとヒトや他の生物で機能が分かっていたタンパク質をコードするような遺伝子をヒトゲノム上で同定したり、又は遺伝子発現の増減を手掛かりに、キーとなる遺伝子産物を精製して調べたり、又は培養細胞に遺伝子を導入することで遺伝子産物を大量に産生したりして機能を一つ一つ調べるという、地道な方法に基づいていた。
そして、そのような努力により、これまでに機能が実験等により確認されているヒト遺伝子は、先述の約2万から2万2千個の遺伝子のうち約5分の1だとされている。なおその中には疾病の研究等により変異を起こすことによる機能の変化について解明されたものもあるが、網羅的というにはほど遠い。
(3)構造解析による遺伝子産物の 解析
上記のように、現象に基づく手さぐり的な遺伝子の機能解明は非効率的だという考えもあり、遺伝子産物であるタンパク質側から、遺伝子産物を網羅的・体系的に調べていくプロジェクトも出てきた。
ヒトゲノム計画の終了時期近く、日本では2002年度から5年間「タンパク3000プロジェクト」が開始された。いわゆる「ポストゲノム」である。それにより、タンパク質の結晶化とX線構造解析、及び核磁気共鳴(NMR)による構造解析によって、大量のタンパク質の基本構造(パターン)の決定が行われた。さらに2007年度より5年間、解明困難な重要タンパク質を対象に「ターゲットタンパク研究プログラム」が行われた。
米国でも同様なプロジェクトが行われ、国際競争のごとく、各種タンパク質の構造が急速に解明されていった。
ただし、それらにより遺伝子の産物であるタンパク質の基本構造は分かったが、それがどのような機能を果たしているかまでの正確な予測はできなかった。
(4)AIによる遺伝子産物の解析
遺伝子産物であるタンパク質の構造・機能解明について、大きな進展をもたらしたのはAIの登場だった。
ディープマインド社は、AIソフト「アルファフォールド(AlphaFold)」を用いて、膨大な既知のタンパク質構造とアミノ酸配列のデータを学習させ、決定に数年かかっていたタンパク質の構造を、数十分で非常に精度よく予測できるようにした。アルファフォールド2ではいっそう進化し、予測されたタンパク質構造数は2024年末の段階で2億を超え、当然ながらヒトゲノム中の2万~2万2千個の遺伝子の産物もそれに含まれていた。彼らはこの業績により、2024年度のノーベル化学賞を授与された。(2024年ノーベル賞特集2 タンパク質の構造予測や設計を行うAIの開発者にノーベル化学賞)なお、この方法により、遺伝子が変異した場合、それによるタンパク質の微妙な構造変化の予測がある程度可能となった。
その後も、こうしたタンパク質の構造、さらには機能予測を持つAIモデル開発は進化し続けている。以下はその代表例である。
・タンパク質言語モデルESM3:エボルーショナリ―・スケール(Evolutionary Scale)社が約28億のタンパク質のデータを学習して開発したモデル。タンパク質の配列・構造・機能の3つを同時に推論し、自然界にはない新しいタンパク質を生成することも可能とした。
・タンパク質デザインモデルBindCraft:スイス・ローザンヌ工科大学を中心に、MITやオランダの研究チームが連携する国際チームが、AlphaFold2の技術を応用し、標的タンパク質の特定領域に結合し、その機能を制御するタンパク質を、従来よりも好成功率で設計できるモデル。
ただし、こうしたタンパク質の構造・機能予測が適用されるのは、あくまで遺伝子産物としてのタンパク質だった。先述のようにヒトゲノムは遺伝子以外の部分が大部分を占めている。また、RNAとしてのみの機能を持つものもある。そうした部分の役割や、ましてや、そのような部分に起こる変異のもたらす影響については、上記のようなタンパク質に対するモデルで推測することはできなかったのである。このため、DNA変異にも対応したゲノム全部位の機能予測のモデルが必要とされてきた。
3.今回のモデルについて
(1)アルファゲノムの特徴
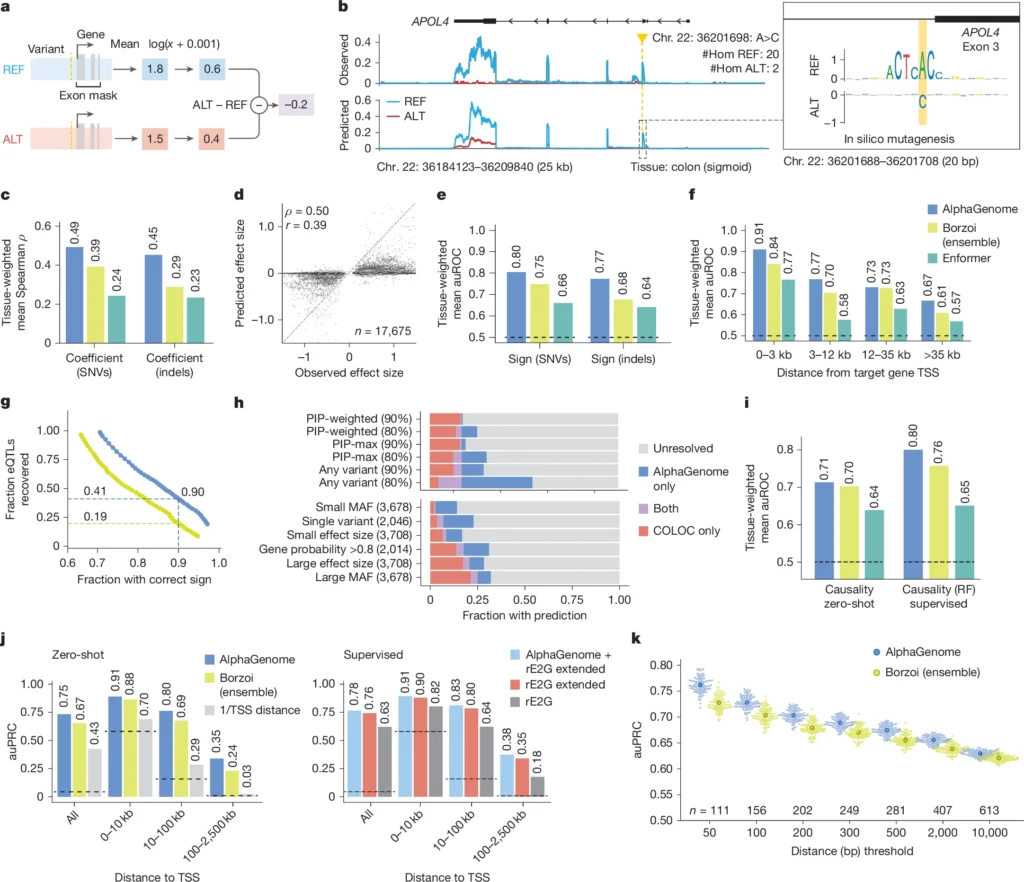
今回、ディープマインド社は、ENCODEやGTExなど、ヒト及びマウス由来の大規模な公開データセットでトレーニングすることにより、アルファゲノムという大規模な予測モデルを構築した。
アルファゲノムは、長大なDNAシーケンスを一度に解析し、しかも遺伝子発現に関する情報(RNA-seq、CAGE、PRO-cap等)のほか、スプライシング部位、転写因子の結合箇所、クロマチンの立体構造など、ゲノムに関わる11種類の主要な機能を同時に予測できる統合モデルである。たとえば、ノンコーディング領域においてある塩基が変異したら、ある遺伝子のRNAのスプライシングがどう変わるか、発現量がどう変わるか、クロマチンの状態はどう変わるか等が、一目で分かるようにした。
従来の手法としては、発現量予測専用モデル、スプライシング予測専用モデル等があったが、それぞれ対象とする現象ごとにモデルを使い分ける必要があった。しかしこのアルファゲノムでは、単一モデルで網羅的なゲノム機能の予測を行える。
また、従来の手法では、一度に解析できる配列の長さは数万~数十万塩基対が限界とされていた。しかも解析する配列の長さが長くなればなるほど予測精度が低くなるという負の相関性があった。だが、アルファゲノムでは、最大100万塩基対という長いDNA配列について高解像度の予測を行える。
特に本モデルが優れているのは、DNA配列上で遠く離れた領域同士が影響しあう遠隔制御の解析である。上述のように、本モデルでは一度に長い配列の解析を行えるため、従来の短い配列の解析ではとらえきれなかったエンハンサーとプロモーターの相互作用等、遠隔制御に係る情報を扱える。
また、本モデルでは1次元のDNA配列情報のみから、「3次元クロマチン接触パターン(コンタクトマップ)」を予測できる。遺伝子の発現調節は、DNAが核内でどのように折り畳まれているかという立体構造と密接に関係している。配列情報からDNAとクロマチンの接触状況を推定できるこの機能により、ゲノム制御機構の解明につながる。
ということで、論文によると、アルファゲノムは、変異効果予測の26の評価項目中25項目において、既存の最先端モデルと同等かそれ以上の性能を示したとのことである。
(2)アルファゲノムの共用
アルファゲノムのソースコードはオープンソースとして公開されている。ライセンスにはアパッシェライセンス(Apache License)2.0が採用されており、商用利用も含め利用や改変、再配布が認められている。
ただし、モデルの学習済み重みパラメータについては非商用の研究目的に限定して提供されており、利用時には別途定められたモデル利用規約への同意が必要となっている。
また、アルファゲノム自体は一般公開されていないが、ディープマインド社はそれを非営利の研究者コミュニティに向けて提供しており、研究目的での利用は基本的に無料になっている。
(3)アルファゲノムの研究・医療への意義
アルファゲノムは、今後、研究や医療にとってどのような意義をもつか。
研究面では、現在はヒトとマウスのデータで訓練されたものであるが、今後、各種生物のデータも追加・訓練することで、各配列のより普遍的な働きが明確化できたり、進化に伴うゲノムの変遷等の研究に役立つ可能性は大きい。
医療面では、ノンコーディング領域について、変異が遺伝子発現やスプライシングなどに与える影響を予測できれば、疾患への寄与や疾患を防ぐための変異についても理解が進む。特に遺伝子検査においては、こうしたノンコーディング領域の変異は「臨床的意義不明(VUS)」として片付けられてきた場合が多かった。だが本モデルにより、少なくとも追加検査の必要性の判断や、解釈の根拠を補強したりするという用途が期待できる。
4.おわりに
AIモデルの進展により、これまで研究者が地道に長時間かけてやっていた研究が、あっという間に実現できるようになってきた。今回のアルファゲノムなどはまさにそうであり、本来、個々の研究の最終目的となるような成果に、ごく短時間で行きつくことが可能となってきた。このことをもって、中には、自分たちの領域を侵食されると考える者もいるかもしれない。著者としては、研究者はぜひとも、AIモデルをうまく利用することにより、自らの研究成果の検証を行ったり、AIモデルの成果を踏まえ、AIでは現在は不可能な、独創的なその先の研究を行う等、ポジティブな共存を図ってもらうよう期待したい。
参考文献
・Z. Avsec et. al. (2026) “Advancing regulatory variant effect prediction with AlphaGenome”, Nature Vol.649, 1206-1218
・文部科学省ライフサイエンス課「AI for Life Scienceに関する動向」(2025/11/25)
https://www.mext.go.jp/content/20Advancing regulatory variant effect prediction with AlphaGenome251121-mxt_life-000046001_7.pdf
・武村政春「じつは、ヒトのゲノムのうち、遺伝子はたったの1.5%…なんと、一番多いのは「祖先に感染したウイルスの遺伝子の残骸」という衝撃の事実」(2024/10/10)現代ビジネス
https://gendai.media/articles/-/136495
・「100万文字DNAを一度に解析──Google DeepMind、11種のゲノム過程を統合予測するAI「AlphaGenome」」(2026/2/2)Ledge.ai HP
https://ledge.ai/articles/google_deepmind_alphagenome_nature_1mb_genome_prediction
・「【AlphaGenome】Google DeepMindのゲノム解析AIとは?概要・性能・使い方まで徹底解説」(20262/02)WEEL HP
https://weel.co.jp/media/tech-category/google
ライフサイエンス振興財団理事兼嘱託研究員 佐藤真輔
