第25回 完全なヒトゲノムの参照配列「パンゲノム」が公開
国際研究チームは、様々な人種からなる47人分の全遺伝情報を高精度で解読したと発表した。今回はその内容や背景等について述べる。
ヒトゲノムの解読については、約1年前の第2回のニューズレターでも紹介したが、もう一度、その進展・精緻化の経緯を簡単に振り返ってみる。
まず第一歩となったのは、1990年から2003年に行われたヒトゲノム計画で、米日英仏独中の6か国の研究機関が参加し、初めてヒトのゲノム解読、つまりヒトの22本の常染色体+XYの性染色体を作る30億塩基対の配列の決定が行われたことである。
それにより、まずヒトゲノムの大雑把な塩基配列(ドラフト)が2000年6月に発表された。これは各国で分担して読んだ配列をとりあえずつなげたというものだった。その後、それを精査してまとめた配列が2003年4月に発表された。通常、ヒトゲノム解読が完了したというと、この発表を指す。
その後の個別のヒトゲノムの解読は、この最初に解読されたゲノムを標準とし、それを参照して比較することで行われてきた。このようなゲノム配列を参照ゲノム(リファレンスゲノム)と呼ぶが、その後、技術の向上等により、ヒトゲノムの新たな領域の発見や、参照配列の間違いの発見等に伴い、この参照ゲノムは定期的に更新されてきた。
それでも解読としては完全ではなかった。その流れをくむ新しい参照ゲノムでも、ゲノム全体の6.7%は読めていなかった。その大きな原因として、反復配列の多い、テロメア(染色体の端の部分)やセントロメア(染色体のほぼ中央に位置し、細胞分裂時にそこを中心として染色体分離が起きる)の解読は困難で、残されたままになっていたのである。
しかし、研究者らは不完全な部分を補うべく努力を続けた。2022年3月、今度こそヒトゲノムの解読が完全になされたとしてScience誌に発表された。これが第2回ニューズレターで紹介したものである。30を超える研究機関による国際的な共同研究組織であるTelomere-to Telomere(T2T)コンソーシアムにより、反復配列を含めたヒトゲノムの最初の完全配列が決定されたのである。この配列はT2T-CHM13と呼ばれる。つまり、一切の脱落のない、1つのゲノムの端から端までの本物の配列が決定されたということだった。
同コンソーシアムでは、反復配列を読みこむため、解析のためばらばらに切断したDNAのつながりが分かるように一度に長い塩基配列を読む方法(ロングリード・シーケンスという)を導入した。なお、それまでは参照ゲノムとしてゲノムすなわち22本の常染色体+XYの性染色体の合計24本が使われていたが、T2T-CHM13では父親由来と母親由来の両方の染色体、すなわち46本の染色体の塩基配列が読み込まれ、発表された。
さて、こうしてヒトゲノム解読という仕事は完全に終わったもの考えられてもおかしくはない。しかし、まだ問題は残されていた。それは、こうして読まれたゲノムが人類を代表するものになっているかということだった。
最初のヒトゲノム計画で解読され、参照ゲノムとして用いられてきたゲノムは、由来としてはわずか約20人分のゲノムであり、しかもその大部分はたった一人からのゲノムだった。T2Tコンソーシアムにしても、1人のゲノムを読んだにすぎない。
人々のゲノムは大部分似通っているが、違いもある。だいたい塩基1000個に1個の違いがあるため、父母由来の染色体を区別すると約60億個の塩基配列中、約600万個の違いがある。また、大きなゲノムの欠失、挿入等、構造変異と呼ばれるものもある。そうしものも含めた遺伝子の多様性を多型と言い、それが人の個性につながったり、疾患に関係していたりする。
そのような違いがあるため、参照ゲノムとしてただ1つのゲノムだけを持っているだけでは心許ない。人種や民族により、持っている多型が異なり、それが人種や民族特有の能力差や疾患へのかかりやすさの原因になったりすることも考えられ、遺伝子研究において単一の参照配列を使用すると、適切な分析ができない可能性がある。
このため、こうした人類のゲノムの多様性にまで配慮して解読し、それを1つの地図に表そうという試みがなされてきた。そのようなゲノムのことを「パンゲノム」と呼ぶ。
それでは今回の発表について説明する。これは米国国立衛生研究所(NIH)傘下の研究所の一つである国立ヒトゲノム研究所(NHGRI)が中心となり、世界の14機関間の協力による「ヒトパンゲノム参照コンソーシアム」(HPRC:Human Pangenome Reference Consortium)で行われたものである。Nature誌に関連論文3本が掲載された。

これは世界の47人の多様な遺伝的背景の人々の全DNAを1つの巨大な遺伝子地図帳にまとめたものである。47人の内訳としては、アフリカ大陸が51%で最も多く、アメリカ大陸34%、アジア13%、ヨーロッパ2%だった。
残念ながら日本人は含まれていないが、これは、今回の解析試料として用いたゲノムの多くが「1000人ゲノムプロジェクト」で用いられたものだということにも起因する。同プロジェクトは2008年に別の国際コンソーシアムが開始したプロジェクトで、世界中の1,000人以上のヒトゲノムを解読して、多様な集団におけるゲノム変異のカタログを作る目的で行われている。同プロジェクトには日本の機関は参加していない。
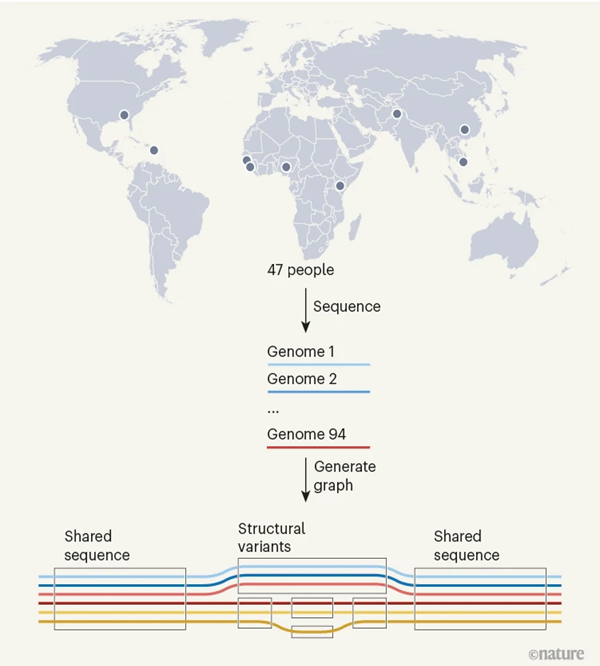
T2Tコンソーシアムで決定した完全なゲノム配列は今回のパンゲノムに組み込まれており、その他の試料についても、完全なゲノム配列を決定するため、ロングリード・シーケンスを用い、またそれぞれについて父母由来の計46本の染色体(つまりゲノム2セット分)の塩基配列を決定した。47人分なので合計94セットのゲノム配列を完全に解読したことになる。
これにより、T2Tの前までの参照ゲノムで欠けてい約1億1,900万塩基対が明らかになり、それは地下鉄路線図のような地図に表わされた。このパンゲノム参照配列を使用すると、従来の参照ゲノムを使用した場合と比べ、変異箇所での読み取りエラーが34%少なくなり、特に反復配列部位でのエラーが減った。また構造変異については1人当たり2倍特定できたとのことである。
つまり、このパンゲノムにより、研究者や医師のゲノム解析時に、いわゆる「参照バイアス」、つまりデータ解析の基礎となる参照ゲノムの影響が抑えられた、より完全なリソースが得られたことになり、ゲノムと病気の関係を調べる分析の精度がより高まったことになる。そうして、従来の研究では観察が困難だった、進化にとっての重要箇所や構造変異等の違いもより明確になると思われる。また、親から受け継いだ染色体をよりよく区別できるようになると思われる。
なお懸念がないわけではない。ゲノム公開に伴う倫理問題である。1990年代に行われたヒトゲノム多様性プロジェクトや、現在米国で進行中のAll of US研究プログラムでは、米国を拠点とする民族グループから、DNAを採取する者から十分同意を得られていないという批判を受けた。その意味では、今回の解析が依存している1000人ゲノムプロジェクトでは、少数民族のものも含め、開始の何年も前に収集された試料も含まれている。
1000人ゲノムプロジェクトの参加者が署名した同意書は、参加者の試料の再分析も認めている。さらに、今回のパンゲノムのプロジェクトに用いる際には、倫理的な収集と遺伝データの使用を確実にするために追加的な措置が講じられ、その過程で、試料提供を拒否する方針を持つ先住民やその他の民族グループに属する人々を含めないことを約束したとのことである。ただ、そうした懸念を払拭できないことも考え、今後は新たな研究参加者を募集することも求められている。
本コンソーシアムの今後の目標としては、2024年半ばまでに少なくとも350人の遺伝的多様性をとらえることである。それによりゲノム研究や医療へのさらなる貢献につながることとなると思われる。日本も東北メディカル・メガバンク計画などで行われている日本人の参照ゲノムの開発に際し、これとも十分連携をしていくことを期待したい。
(参考文献)
・A. Massarat & M. Gymrek (2023) “A collective human reference genome”, Nature; Vol.617, 256-258
・L. Liverpool (2023) “First human ‘Pangenome’ aims to catalogue diversity”, Nature; Vol.617, 444-445
・ジャンルカリッチョ「遺伝学の大きな飛躍:パンゲノムの最初の草案」FUTUROPROSSIMO(2023/5/12)(https://ja.futuroprossimo.it/2023/05/un-grande-passo-per-la-genetica-la-prima-bozza-del-pangenoma-umano/)
ライフサイエンス振興財団嘱託研究員 佐藤真輔
